
CE MDD 의료기기
의료기기 지침은 유럽 연합 내에서 의료기기에 관한 법률을 조화하기 위한 것입니다. 법적으로 유럽 시장에 의료기기들을 판매하기 위해서는 제조자들은 MD지침(Medical Device Directive)의 요구 사항을 준수해야 합니다. 신청자의 제품 및 품질 시스템을 평가해야 하며 제조자는 제품들을 판매 하기전에 CE 마크를 부착해야 합니다.
-
-
 < 적용 품목 >
< 적용 품목 >
-
적용 품목
의료기기 지침은 '의료기기'의 정의에 따르는 장치에 적용 가능합니다. 의료기기란 제조자의 의도대로 인간에게 사용되는 목적을 가진 소프트웨어를 포함하는 단독 또는 조합으로 사용되는 기계, 장치, 기기, 재료 또는 기타 물품입니다.
진단, 예방, 감시, 치료 또는 질병의 경감, 진단, 감시, 치료, 상해 또는 장애에 대한 완화 또는 보상, 조사, 교체 또는 해부학적 또는 생리학적 과정의 수정, 개념의 제어, 약리적, 면역적 또는 신진대사적 수단에 의해 인체 내에 또는 인체상에 의도한 주요 작용을 달성하지는 않지만 그런 수단에 의해 그 기능을 도와줄 수 있는 것을 말합니다.
-
-
-
적격성 평가 절차
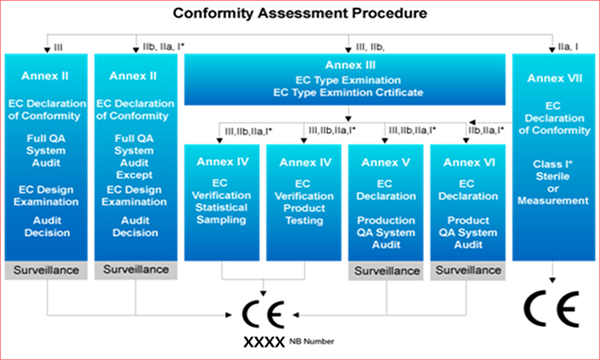 < 적격성 평가 절차 >
< 적격성 평가 절차 >
-
분류법
의료기기 지침서(93/42/EEC)의 부속서 IX는 의료기기를 분류할 수 있는 18개의 규칙을 제공합니다. 이 규칙들에 따라 의료기기는 의료기기의 의도된 목적에 따라 분류됩니다.
- Rule 1 ~ 4 : Non-invasive 기기
- Rule 5 ~ 8 : Invasive 기기
- Rule 9 ~ 12 : 능동형 기기
- Rule 13 ~ 18 : 특별법
환자 및 사용자에 대한 의료기기의 위험 평가는 위의 18개의 규칙에 따라 식별되고 분류됩니다. 식별된 위험 값을 바탕으로 적격성 평가 절차가 결정됩니다. 위험 값이 높을수록 적격성 평가 요구 사항이 엄격 해집니다.
Class I 의료기기는 부속서 VII의 평가 절차를 따릅니다.
-
Class I의 절차 (측정 기능 포함)
제조사의 선택 가능 사항 :- 부속서 VII 섹션 3 및 부속서 IV에 따른 각각 또는 통계적으로 선택된 샘플에 대한 도량형에 관한 요구사항을 만족하는 적격성의 검증 및 기술 문서화
- 부속서 VII 섹션 3 및 통계학적 요구 사항과 연관된 부속서 V에 의한 제품 품질 시스템의 평가 및 기술 문서화
- 부속서 VII 섹션 3 및 통계학적 요구 사항과 연관된 부속서 VI에 의한 의료기기의 품질 평가 및 기술 문서화
-
Class I의 절차 (멸균 포함)
제조사의 선택 가능 사항 :- 부속서 VII 섹션 3 및 멸균 상태와 연관된 부속서 V에 의한 제품 품질 시스템의 평가 및 기술 문서화
-
Class IIa 의료기기 절차
제조사의 선택 가능 사항 :- 부속서 VII 섹션 3 및 부속서 IV의 섹션 8에 의한 각각 또는 통계학적으로 선택된 샘플에 대한 적격성 검증 및 기술 문서화
- 부속서 VII 섹션 3 및 부속서 V의 섹션 6(제조사의 현장에서 심사 된)에 의한 제품 품질 시스템의 평가 및 기술 문서화
- 부속서 VII 섹션 3 및 부속서 VI의 섹션 6(제조사의 현장에서 심사 된)에 의한 의료기기 품질의 평가 및 기술 문서화
- 부속서 VII 섹션 3 및 부속서 II(섹션 4에 따라 개발 평가가 제외 된)에 의한 전체적인 품질 시스템이 제조자의 현장에서 심사 된 평가 및 기술 문서화
-
Class IIb 의료기기 절차
제조사의 선택 가능 사항 :- 부속서 III 섹션 3 및 제조된 각각에 대한 적격성 검증 및 유형 검사 와 기술 문서화 : 부속서 IV에 의해 통계학적으로(무작위로) 선택 된 샘플이나 섹션 5에 의해 검증 되어야 합니다.
- 부속서 III 섹션 3 및 부속서 V(제조사의 현장에서 심사 된)에 의해 제조 품질 시스템의 평가 및 유형 검사와 기술 문서화
- 부속서 III 섹션 3 및 부속서 VI(제조사의 현장에서 심사 된) 섹션 6에 의해 의료기기 품질의 평가 및 유형 검사의 기술 문서화
- 부속서 II 섹션 3.2 및 부속서 II(섹션 4에 의해 개발 평가가 제외된)에 의해 제조사의 현장에서 심사가 이루어진 모든 품질 시스템의 평가에 대한 기술 문서화
-
Class III 의료기기 절차
제조사의 선택 가능 사항 :- 부속서 II 섹션 3.2 및 부속서 II(제조사의 현장에서 심사된)에 의한 모든 품질 시스템의 평가 및 섹션 4.2에 의해 개발의 설명에 대한 기술 문서화
- 부속서 III 섹션 3 및 제조된 각각에 대한 적격성 검증 및 유형 검사의 기술문서화 : 부속서 IV 섹션 6에 의해 통계학적으로(무작위) 선택된 샘플에 대한 섹션5에 의해 검증이 이루어져야 합니다.
- 부속서 III 섹션 3 및 부속서 V 섹션 3.2에 의해 제품 품질 시스템(제조사의 현장에서 심사된)의 평가 및 유형 검사의 기술 문서화
-
-
-
-
 < MDD 종료 시점 >
< MDD 종료 시점 >
-
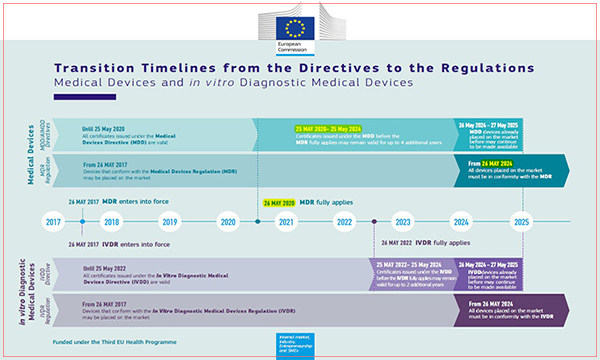
MDD 종료 시점
유럽의 의료기기 지침인 MDD (93/42/EEC)는 2020년 5월 26일부터 MDR로 완전히 대체됩니다. 이후에는 MDD로의 CE인증 신청이 불가능하며, 새로운 의료기기는 MDR (2017/745/EU) 요구사항을 충족시켜야만 합니다.
하지만 강제적용 시점 전에 MDD의 적합성 평가를 마무리하여 인증서를 발급받은 경우, 최대 2024년 05월 27일까지 인증서의 효력을 인정받아 제품을 유럽 관할구역내에 출하할 수 있습니다.
유럽의 Competent Authority에서 공식적으로 발행한 상기의 자료를 확인해 보실 수 있습니다.
-
-
-
 < CE / MDD 제공 서비스 >
< CE / MDD 제공 서비스 >
-
CE / MDD 제공 서비스
IGC는 현재 3개의 Notified Body(이하 NB)기관과 협력하여 CE/MDD 프로젝트를 진행하고 있습니다. 이로 인해, Class l 부터 Class lll까지 거의 모든 스콥에 관하여 예외 없이 신청을 진행할 수 있습니다. 수많은 프로젝트를 진행하며 쌓은 경험을 토대로 귀사의 CE/MDD 인증 획득을 위하여 지원을 아끼지 않겠습니다.
-
우리가 제공하는 관련 서비스
01경영시스템 인증
02제품인증 서비스
03제품 시험
04심사자격 인증 서비스
05심사원 교육 및 양성